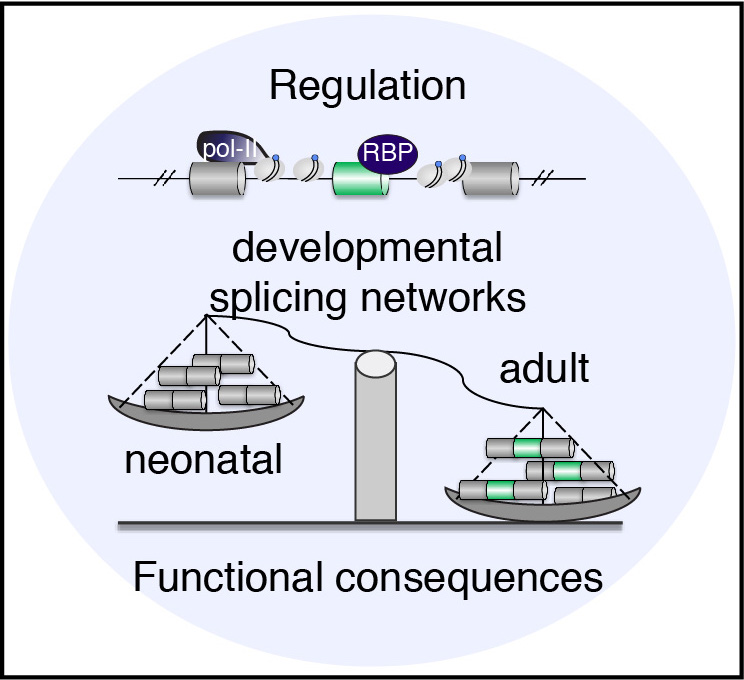
 Alternative splicing is a posttranscriptional mechanism that explains how individual genes can produce more than one transcript due to the inclusion or exclusion of specific regions originating multiple protein isoforms with diverse features. More than 90% of human genes undergo alternative splicing. During development multiple transcriptional and post-transcriptional networks are coordinately regulated to drive organ maturation, tissue formation, and cell fate.
Alternative splicing is a posttranscriptional mechanism that explains how individual genes can produce more than one transcript due to the inclusion or exclusion of specific regions originating multiple protein isoforms with diverse features. More than 90% of human genes undergo alternative splicing. During development multiple transcriptional and post-transcriptional networks are coordinately regulated to drive organ maturation, tissue formation, and cell fate.
A wide variety of human diseases are caused by mutations within cis-acting sequences of pre-mRNAs that produce aberrant alternative splicing and alter function of the mutated allele (Duchenne muscular dystrophy, Cystic fibrosis, Limb girdle muscular dystrophy 1B, among others). In addition, certain splice isoforms are depleted or induced in cancer, diabetes, or muscular dystrophies suggesting specific physiological roles. Defects in the function or expression of RNA binding proteins and core spliceosome constituents induce splicing mis-regulation in multiple severe diseases.
Tissue specific alternative splicing is critical to maintain tissue identity. Brain, heart, and skeletal muscle are the tissues where the most tissue specific splicing takes place raising the questions of how developmental stage- and tissue-specific splicing influence protein function and how this regulation occurs. To investigate this big picture question, our research program has three main angles:
1) the molecular mechanisms that explain how alternative splicing regulates the expression of trafficking and membrane dynamics proteins in normal development and diseases. We are interested on identifying the regulators of the trafficking splicing networks (RNA binding proteins, epigenetic’s contributors). The ultimate goal is to understand how these developmental networks are normally coordinated in development. In this manner, we expect to provide new knowledge of how these networks are uncoordinated in diseases.
2) how alternative splicing impacts on the functions of membrane trafficking proteins in internal cell architecture and physiology. We are identifying the functional impact of splicing regulation of membrane trafficking proteins. Furthermore, we aim to elucidate the full mechanisms linking those physiological functions with the splicing isoforms.
Angles 1 and 2 are initially focused on striated muscles (heart and skeletal muscle) because we found that trafficking and membrane dynamics genes are regulated by splicing specifically in these tissues and some of them also in brain. Misregulation of membrane trafficking proteins is one hallmark of muscle and brain disorders. In particular, striated muscle cells have contractility primordial functions that require a very precise formation and maintenance of internal architecture through membrane invagination and trafficking processes. Receptors and ion channels need to be correctly transported to these locations to exert their key functions. We are studying the crosstalk between alternative splicing and membrane trafficking and its contribution to the maturation and maintenance of the myocyte exquisite architecture and functions.
3) the roles of epigenetic dynamics on alternative splicing regulation in postnatal striated muscle development. The contribution of epigenetics in alternative splicing has been very well documented and demonstrated in different cell culture systems. However, this epigenetic – alternative splicing interplay has not been studied within developmental or disease contexts. Our hypothesis is that during muscle development, epigenetic dynamics triggers specific histone marks around the alternative spliced exons altering RNA Polymerase II kinetics, which in turn favors the recognition of weak splice sites and thus inclusion of alternative exons or recruitment of specific splicing factors that promote exon inclusion or skipping.
In the lab, we utilize a broad spectrum of approaches ranging from molecular biology, cell biology, microscopy, deep-sequencing, and physiology techniques. We use cell culture systems and animal models.
